A standard for aseptic filling or a rare necessity?
What is PUPSIT?
In recent years, much discussion was raised on the need for pre-use/post-sterilization integrity testing (PUPSIT) of sterilizing filters to detect nonintegral filters before they are used for aseptic filling. The reasoning behind PUPSIT vs. a single post-use filter test is that there might be a possibility that a filter passing the post-use test could have allowed bacterial penetration during filtration of the drug product. This phenomenon is referred to as “filter flaw masking”; meaning that the damage of the filter, caused by for example the sterilization step, allows bacterial penetration, but becomes plugged during the filtration process to such extent that it allows the filter to pass the post-use test. To allow this masking phenomenon, two conditions must exist:
1) There must be a defect present in the filter that is big enough to allow bacterial penetration, yet small enough to become clogged during filtration;
2) The product being filtered must be capable of blocking that defect to such extent that the filter passes the post-use test.
Although the idea of PUPSIT seems rational, it also comes with a lot of drawbacks which might outweigh the benefits.

Figure 1: Examples of sterilizing filters (adapted from www.merckmillipore.com)
Annex 1 on PUPSIT
The Annex 1 of 2008 states the following with regard to before pre-use testing:
“113. The integrity of the sterilised filter should be verified before use and should be confirmed immediately after use by an appropriate method such as a bubble point, diffusive flow or pressure hold test. The time taken to filter a known volume of bulk solution and the pressure difference to be used across the filter should be determined during validation and any significant differences from this during routine manufacturing should be noted and investigated. Results of these checks should be included in the batch record. The integrity of critical gas and air vent filters should be confirmed after use. The integrity of other filters should be confirmed at appropriate intervals. “
Because of the wording, it was easy for sterile manufactures to genuinely feel compliant with regard to the pre-use testing requirements by testing the filter offline before use and thus before sterilization. Also in case of single use filtration systems which are gamma radiated they could consider themselves compliant by taking the filter supplier’s certification with regard to pre-use testing. However, this certification does not cover any damages that are introduced during the shipment process, receiving and storage by the end user, as well as handling and installation by the end user. The recent 2022 update of Annex 1 however does not leave much room for interpretation, although they also recognize that use of PUPSIT is not always feasible in which an alternative approach in the form of a risk assessment is allowed:
“8.87 The integrity of the sterilised filter assembly should be verified by integrity testing before use (pre-use post sterilisation integrity test or PUPSIT), to check for damage and loss of integrity caused by the filter preparation prior to use. A sterilising grade filter that is used to sterilise a fluid should be subject to a non-destructive integrity test post-use prior to removal of the filter from its housing. The integrity test process should be validated and test results should correlate to the microbial retention capability of the filter established during validation. Examples of tests that are used include bubble point, diffusive flow, water intrusion or pressure hold test. It is recognized that PUPSIT may not always be possible after sterilisation due to process constraints (e.g. the filtration of very small volumes of solution). In these cases, an alternative approach may be taken providing that a thorough risk assessment has been performed and compliance is achieved by the implementation of appropriate controls to mitigate any risk of a non-integral filtration system.”
Furthermore, they also specify different points which manufacturers need to consider in such risk assessment. These include process knowledge of the specific product type and its potential to alter integrity testing values, but also in-depth knowledge and control of the filter supply chain and sterilization process.
Flaw masking studies
To determine if and under which conditions the hypothesized masking of filters can occur, masking studies were performed. These studies allow the industry to better understand filter masking and thus provide the knowledge needed to accurately perform the required risk assessments as mentioned in the recent update of Annex 1.
In these studies worst-case scenarios were challenged, being marginally flawed cartridge filters (obtained from filter manufacturing lines) presented with proteinaceous solutions to plug the defects and create a passing post-use filter test. Keep in mind that these conditions are rare as most filter rejects are catastrophic failures (large defects that make the filter unable to be integrity tested); moreover, marginal failures may still completely block bacterial passage. The results show that out of 24 tested filters, challenged with 24g/L foulant concentration and 90%+ flow decay, only 2 filters demonstrated “flaw masking” (= failed pre-use test and passed post-use test). In another set of experiments, disc filters with defects created via laser-drilling of 10µm holes were used. For these filters, an automated integrity tester was able to detect all damaged filters at any blockage level up to 75%.
The data obtained by these masking studies clearly show that, although there is a change of masking, it is not likely to occur under standard drug manufacturing conditions. Only fluids with unusually high foulant concentrations in combination with atypical use of filters (e.g. long filtration times), might lead to levels that approach high blockage conditions and an increased risk of masking. However, this most likely will also result in an excessive flow decay which is not feasible. Therefore, assessment of product characteristics and filter flow decay are crucial parameters for manufacturers in their evaluation for the need of PUPSIT.
Bacterial Challenge Test data mining study
The minimal risk of filter masking in the pharmaceutical industry was also demonstrated by a Bacterial Challenge Test (BCT) data mining study. This study relied on the concept that any fluid with a potential clogging mechanism, should also result in an increase of the bubble point value due to excessive pore plugging. In other words, the ratio between the bubble point value of the post-use and pre-use test can indicate whether a flaw-masking fluid is present.
To translate this concept to the industry, the authors used the historical integrity test results from over 2,000 filters used in BCTs to evaluate whether a bubble point inflation mechanism – and thus risk for flaw masking – exists for any given fluid and filter combination. Similar to the flaw masking studies, the results from these BCTs are worst case as the sterilizing grade (0.2 µm) filters are being exposed to product, plus a high concentration of the bacterial challenge organism which contributes to the plugging of pores and potential flaws of the filter. Also parameters such as duration of filtration, volumes and flow rates are usually worst case during BCTs compared to the process being validated. In parallel with the BCT of the sterilizing filters, a filter with a larger pore size (usually 0.45 µm) is usually being challenged as positive control to confirm that viable and sufficiently small bacteria are used and are able to penetrate the filter. These filters, which are also tested pre -and post-use, will have lower bubble point results due to the larger pores and can be considered as a model for marginally flawed filters. If the 0.45 µm filter becomes so fouled that its post-use test result exceeds the minimal passing value of the 0.2 µm sterilizing filter, flaw masking would be a risk for this fluid/filter combination.
The outcome of the data mining study showed that the mean ratio of the post-use to pre-use bubble point test was 1.00 ± 0.15, indication that on average there is no trend of bubble point inflation within the industry. When looking at individual fluid/filter combinations, the data mining team determined that only a small fraction (1.5%) of the filter test conditions demonstrated ratios that indicate a theoretical risk towards flaw masking. Despite the low percentage, the actual risk is probably even lower as the BCTs are performed under worst-case conditions and with the addition of bacteria. Furthermore, this study also shows that the BP ratios observed during routine bacterial retention testing is one means to assess the risk for masking of specific fluid/filter combination and thus can be considered when determining whether a PUPSIT should be implemented.
Drawbacks of PUPSIT
As mentioned earlier, there has been a lot of discussion with regard to the implementation of PUPSIT as it may also – and this might sound quiet controversy – add risk to the sterile manufacturing process. The main drawback of incorporating PUPSIT into a filtration process is the increase in complexity of the process, and especially for redundant filtration systems (see Figure 2). This increase complexity comes with additional risks and drawbacks which need to be taken into account before implementing PUPSIT. The most common drawbacks involve:
- Performance of filter integrity test in line requires the system to maintain much higher pressures, often exceeding 60 psi. This increases the risk for sterile boundary leaks, especially when using single-use equipment which often involve weaker tubing junctions/valves compared to stainless steel piping. Also the increased amount of valves and flushing bags/containers adds to this increased risk for leaks.
- Wetting of the filter with buffer, required to perform the pre-use test, could result in dilution of the product; unless the filter is dried or the first volume is discarded
- Much longer process times as extra steps are required such as filter wetting, blow drying, and the need to adapt to non-standard situations such as re-wetting the filter. Moreover, there is the need to test all additional protective vent filters which come with PUPSIT systems.
- The need for system manipulation (e.g. opening/closing of valves) on the sterile side of the filter, making the PUPSIT system more vulnerable to human errors. Although this risk can be eliminated via the use of automated systems.
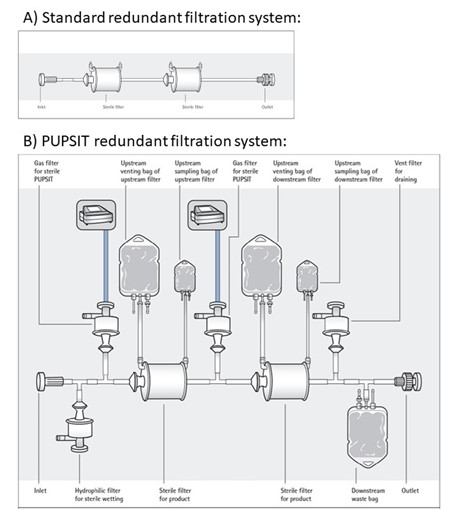
Figure 2: Schematic overview of non-PUPSIT (A) and PUPSIT (B) redundant filtration systems, illustrating complexity of PUPSIT implementation (source: www.pda.org)