Introduction
As described in cGMP regulations (21 CFR 211.160 & 211.165), laboratory testing of components, containers & closures systems, in-process materials, bulk- and finished products is mandatory to ensure product quality and patient safety.
Tests will be performed as described in previously defined specifications, and if a result falls outside of the specification limits or acceptance criteria, failure is described as an out-of-specification (OOS) result. Obtaining OOS results can impact the release decision and worst-case result in a rejection of commercial batches, leading to inventory loss, financial/economic loss, and reputation/company image damage. But most importantly, it may compromise the safety of patients if not handled correctly.
For every OOS result that is obtained, an investigation needs to be conducted and recorded, including its conclusions and follow-ups (21 CFR 211.192) (1). The aim of the investigation is to determine the root cause of the OOS result, which can be due to:
- An error during the laboratory testing (not product-related);
- An aberration during the manufacturing process (product-related).
Once the root cause has been detected, it can be determined if there is also impact on other batches of the same drug product or other products. In conclusion, an investigation of an OOS result is always necessary, even if the batch will be rejected. In below sections, a solid framework for this specific type of investigations is provided.
OOS Investigation steps (cfr. FDA & MHRA)
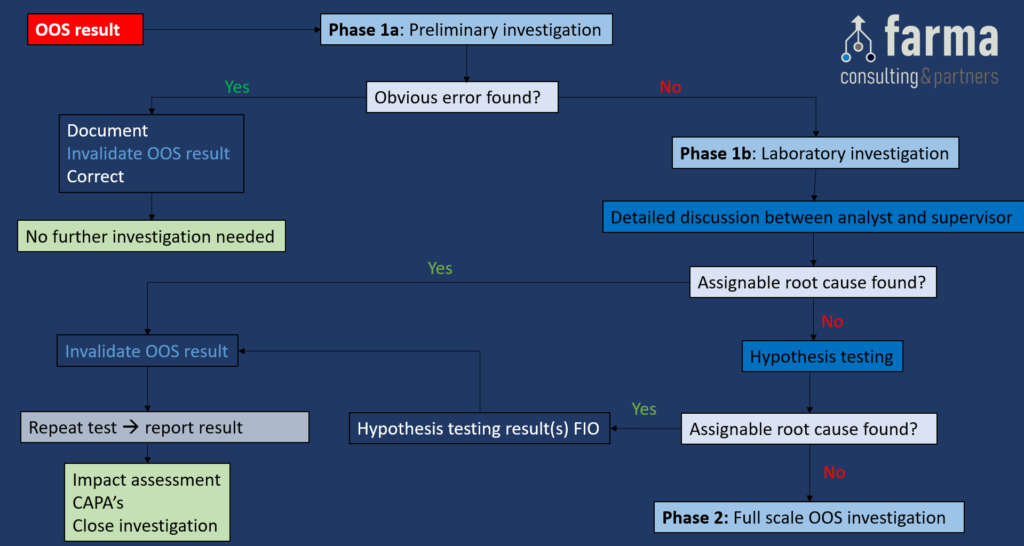
Phase 1a: Preliminary investigation (responsibility of the Analyst)
Objective: Determine if an obvious lab error has occurred.
For example: calculation error, incorrect instrument parameter, spiling of sample solution, etc.
Depending on the outcome of the preliminary investigation, the following actions can be taken:
- If an obvious error has been detected, the analyst should immediately document what happened, invalidate the OOS result, correct it, and no further investigation is required.
- If no obvious error has been found, GO TO Phase 1b.
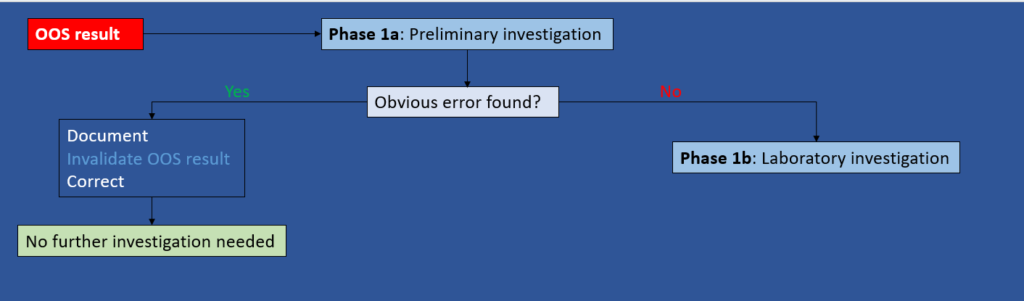
Figure 1: Phase 1a – Preliminary Investigation
Phase 1b: Laboratory Investigation (responsibility of the Supervisor)
Objective: A more thorough investigation with the focus on laboratory procedures and processes with the goal of finding an assignable root cause for the OOS result.
This step can consist of the following topics:
- Detailed discussion between analyst and supervisor. The use of a Laboratory Investigation Checklist is recommended and can contain – but is not limited to – the following checks:
- Correct test methodology followed?
- Correct sample(s) taken/tested?
- Correct and clean glassware used?
- Review equipment logbooks
- System suitability conditions met?
- Analyst trained on the method?
- Check if other OOS results were obtained for the same product
- Hypothesis testing: These are predefined (2) and pre-approved tests, to confirm or exclude an assignable root cause for the OOS result. The results of the hypothesis tests are only to support the investigation (thus for information only), and cannot be used as a valid test result to be reported.
It is important to note that the hypothesis test plan must be approved before hypothesis testing can occur. Examples of hypothesis tests:- Re-injection of the test solution
- Injection of a new vial from the final dilution
- Injection of newly prepared dilutions from the stock solution
- A new sample preparation may be made and analysed in some situations, for example: to examine the hypothesis of improperly handled initial sample preparation or when a destructive analysis was used (e.g. Karl Fischer). However, QA approval is recommended, since the assignable root cause may remain unfound when a completely new sample is analysed.
Depending on the outcome of the phase 1b investigation, the following actions can be taken:
- If an assignable root cause has been found, invalidate the OOS result, mark the hypothesis results as for information only, and perform a Repeat Test (3). Report the repeat test result, perform an Impact Assessment, define appropriate CAPA’s & close the Investigation.
- If no assignable root cause has been found, GO TO Phase 2.
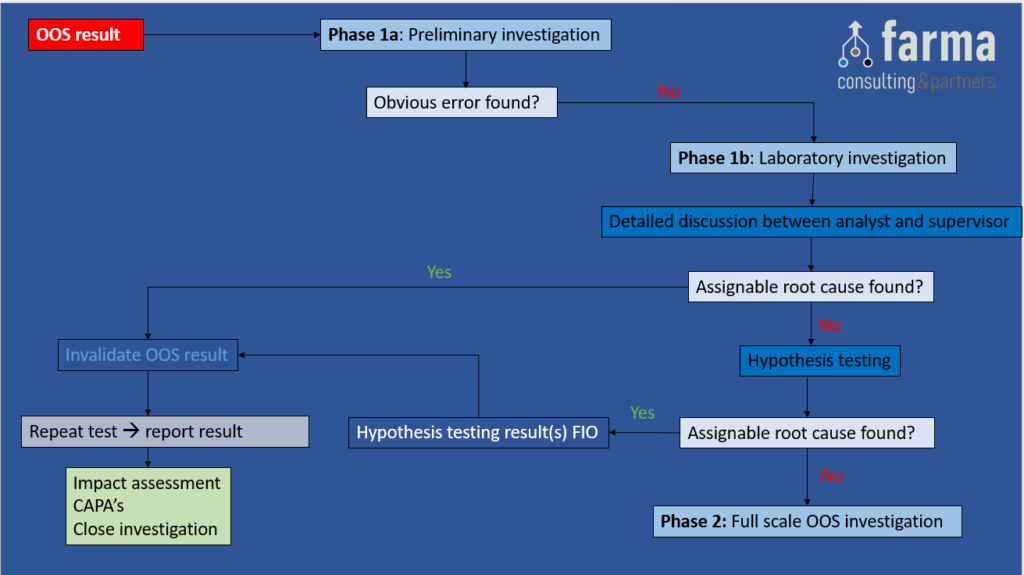
Figure 2: Phase 1b – Laboratory Investigation. A) Discussion between analyst & supervisor and B) Hypothesis testing
Phase 2: Full scale OOS Investigation
Objective: This investigation should be executed conform a predefined procedure, and should include a detailed investigation outside the lab. The investigation should focus on the manufacturing processes to determine whether there is an assignable root cause originating from the manufacturing process
Depending on the outcome of the phase 2 investigation, the following actions can be taken:
- If an assignable root cause originating from the manufacturing process has been found, DO NOT invalidate the OOS result, report the OOS result, raise a deviation/failure investigation (4), define appropriate CAPA’s & close the investigation. In addition, the QP will need to make a decision regarding batch disposition/rejection.
- If no assignable root cause originating from the manufacturing process has been found, GO TO step Additional Laboratory Testing.
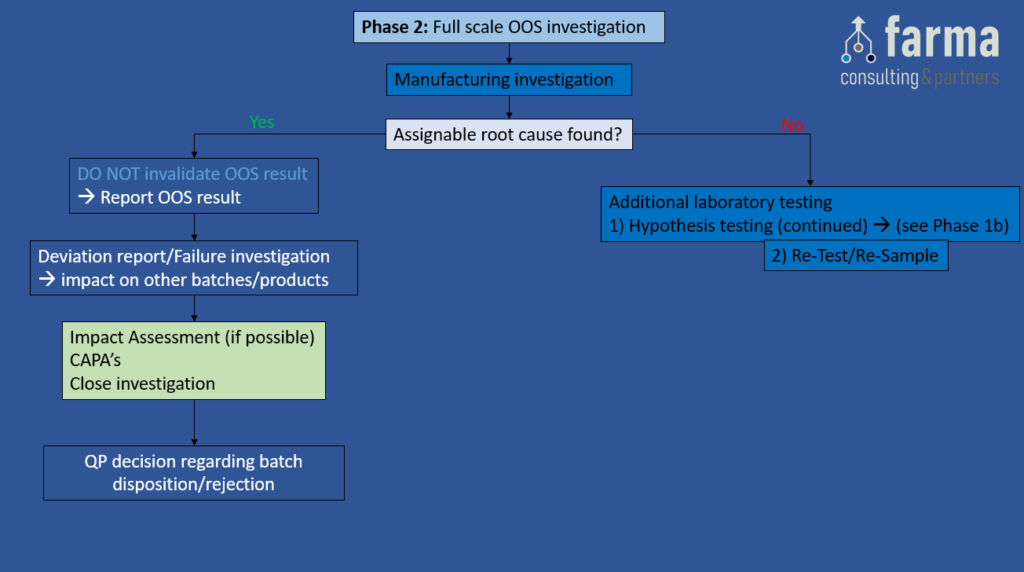
Figure 3: Full scale OOS investigation. A) Manufacturing investigation
Additional laboratory testing
Objective: Perform additional laboratory testing to find an assignable root cause. This can include the following steps:
- Further hypothesis testing (refer to Phase 1b) can be performed.
- Re-testing / Re-sampling (5) can be performed:
- For the re-test, a portion of the original sample is used. The decision on how to re-test and the minimum number of re-tests to be performed, should be described in a procedure, and should be based on scientifical principles.
! The re-test plan must be approved before re-testing can occur. - A re-sample may be used if 1) there is insufficient material remaining from the original sample or if 2) there is a proven issue with the original sample’s integrity (e.g. wrong sample preparation). A re-sample may be used only after QA approval.
! The re-sampling plan must be approved before re-sampling can occur.
If the investigation determines that the initial sampling method was in error, a new accurate sampling method shall be developed, qualified and documented. It’s recommended to perform testing by a different analyst, who is at least as experienced and qualified in the method as the original analyst perform the re-testing/re-sampling.
- For the re-test, a portion of the original sample is used. The decision on how to re-test and the minimum number of re-tests to be performed, should be described in a procedure, and should be based on scientifical principles.
Depending on the results obtained from additional hypothesis testing, retesting or resampling, the following actions can be taken:
- If the re-test/re-sample confirms the OOS result, DO NOT invalidate the OOS result, report all results, perform an Impact Assessment (if possible, based on a most probable root cause), define appropriate CAPA’s & close the Investigation. In addition, the QP should make a decision regarding batch disposition/rejection.
- If the re-test/re-sample doesn’t confirm the OOS result (meaning the re-test/re-sample results are within specifications), invalidate the OOS result, report the (averaged) (6) re-test/re-sample result, perform an Impact Assessment (if possible, based on a most probable root cause), define appropriate CAPA’s & close the Investigation.
Full Flow Chart

Tips & tricks
- Only invalidate the OOS result when there is 1) an obvious error or 2) a laboratory assignable root cause!
- Collaborate with and include all parties necessary: analyst, reviewer, equipment responsible, cleaning responsible, subcontracted laboratories if applicable, etc…
- Don’t discard your test preparations immediately, they may be needed for hypothesis testing.
- Avoid Testing Into Compliance, meaning analyzing a sample many times until a PASS result is obtained and then invalidating the OOS result and reporting the PASS value, without scientific justification.
- Clearly indicate which results are INVALID, For Information Only (FIO) or VALID and will be reported.
- Try to be scientifically correct when drafting up hypothesis- & re-test-plans.
- A lot of new results will be created in scope of the investigation, clearly indicate which result is generated in which testing phase/hypothesis.
- Use tables to clearly represent your data.
Conclusion
Correct management of OOS results in quality control testing is crucial for ensuring product quality and patient safety. By following standardized procedures, identifying root causes, implementing corrective actions, and documenting all steps thoroughly, you can ensure the well-being of patients.
We hope to have provided you with a good framework to handle OOS results. Still facing some questions? Contact us today!
Definitions
Specifications: a list of tests, references to analytical procedures, and appropriate acceptance criteria, which are numerical limits, ranges, or other criteria for the tests described.
Out of specification (OOS) result: a test result that falls outside the specifications or acceptance criteria established in the drug applications, drug master files, official compendia, by the manufacturer etc.
Obvious error: An evident/apparent/clear reason for obtaining an OOS result.
Hypothesis Testing/Re-analysis: Testing performed to help confirm or discount a possible root cause i.e what might have happened that can be tested.
Repeat testing: An additional test, done by the same analyst, to replace the original invalid data when a laboratory assignable root cause has been identified.
Re-testing: An additional test, done by a different analyst, to confirm the original OOS test result when a laboratory assignable cause has not been identified.
References
ICH Q6A guidance on specifications: ICH Q6A specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances – Scientific guideline | European Medicines Agency (europa.eu)
Code of Federal Regulations – 21 CFR Part 211: eCFR :: 21 CFR Part 211 — Current Good Manufacturing Practice for Finished Pharmaceuticals
EudraLex – Volume 4, Chapter 6: GMP_chapter6_final (europa.eu)
Both the FDA and the MHRA provide guidance documents on how to investigate OOS results:
- FDA guidance:
Comparison of the FDA guidance & the MHRA guidance: (14) OOS-FDA vs. MHRA Guidelines | LinkedIn
Pharmablog – SOP for Out Of Specification (OOS) as per MHRA & USFDA Guidelines: Investigating and Handling Of Out Of Specification (OOS) Test Results SOP – PharmaBlog
ComplianceQuest – OOS report : What is OOS (Out of Specification)? -OOS Investigation and OOS Results (compliancequest.com)
GMPSOP : 04 Steps to Investigate Out of Specification (OOS) Result (gmpsop.com)
Difference between OOS/OOT/OOE results:
(1) The EU (EudraLex Volume 4, Chapter 6) also demands that every OOS result is subject to investigation, and this conform a predefined procedure. However, it doesn’t give specific guidance on how to investigate the OOS result, in contrast to the FDA & MHRA.
(2) What needs to be pre-defined:
- Which hypothesis is to test which root cause
- What samples will be tested
- The exact execution of the testing
- How the data will be evaluated
(3) Repeat testing: An additional test, done by the same analyst, to replace the original invalid data when a laboratory assignable root cause has been identified.
(4) The deviation/failure investigations report assesses the impact on other batches or products, that may have been associated with this specific failure.
(5) Re-testing: An additional test, done by a different analyst, to confirm the original OOS test result when a laboratory assignable cause has not been identified.
(6) Averaging test data needs to be done appropriate and can only be done if it’s specified by the test method (meaning it was also done during the original testing that produced the OOS result). This because averaging can hide the variability among individual test results.